Transfer Labels from a Retina scRNA Reference onto a Query Dataset
2023-07-25
morph_scRNA_query_onto_reference.RmdIntroduction
When we discuss data projection (or morphing! branding!), we have two primary kinds of data. First, the reference, which has been previously analyzed and labelled. Second is the query, which is new data that you wish to speed up your understanding of by transferring knowledge (like cell type or tissue label) from the reference.
metamoRph::run_pca
This takes in raw count data, matched meta data, and outputs a list
object containing a prcomp object and some extra
information: percent standard deviation explained by each PC, a
center_scale list which has the center and scale values
calculated in prcomp and are needed to properly normalize
the query data, the meta data itself, and finally the parameters given
to metamoRph::run_pca
library(Seurat)
# library(metamoRph)
library(dplyr)
library(uwot)
library(ggplot2)
tictoc::tic()
# download:
# https://hpc.nih.gov/~mcgaugheyd/scEiaD/2021_11_11/study_level/SRP255195.seurat.Rdata
load("~/Downloads/SRP255195.seurat.Rdata")
reference <- scEiaD # these objects from the plae resource are named 'scEiaD' so we should rename it right away
#### filter down to cell types with at least 50 cells
reference_meta <- reference@meta.data %>%
as_tibble(rownames = "bc")
set.seed(2023.0721)
celltypes_to_keep <- reference_meta %>%
group_by(CellType_predict) %>%
summarise(Count = n()) %>%
filter(Count >= 50, !is.na(CellType_predict)) %>%
pull(CellType_predict)
reference_meta <- reference_meta %>%
filter(CellType_predict %in% celltypes_to_keep)
######## cut down reference matrix to the well represented cell types
ref_mat <- reference@assays$RNA@counts[, reference_meta$bc]
#
mm_pca <- metamoRph::run_pca(feature_by_sample = ref_mat, meta = reference_meta, method = "irlba",
irlba_n = 50)Diversion
You can put these principal components (store in
mm_pca$PCA$x) into UMAP to make a dangerous (distance has
little meaning!) but useful visualization
umap_reference <- uwot::umap(rbind(mm_pca$PCA$x[, 1:20]), ret_model = TRUE)
man_color <- pals::glasbey()
names(man_color) <- celltypes_to_keep %>%
sort()
ref_viz <- umap_reference$embedding %>%
as_tibble(rownames = "bc") %>%
left_join(bind_rows(reference@meta.data %>%
as_tibble(rownames = "bc"), new_data@meta.data %>%
as_tibble(rownames = "bc"))) %>%
ggplot(aes(x = V1, y = V2, color = CellType_predict)) + geom_point(size = 0, alpha = 0.2) +
cowplot::theme_cowplot() + xlab("") + ylab("") + scale_color_manual(values = man_color) + ggrepel::geom_text_repel(data = . %>%
group_by(CellType_predict) %>%
summarise(V1 = mean(V1), V2 = mean(V2)), aes(label = CellType_predict), bg.color = "white") +
theme(legend.position = "none") + ggtitle("Reference: SRP255195")
ref_viz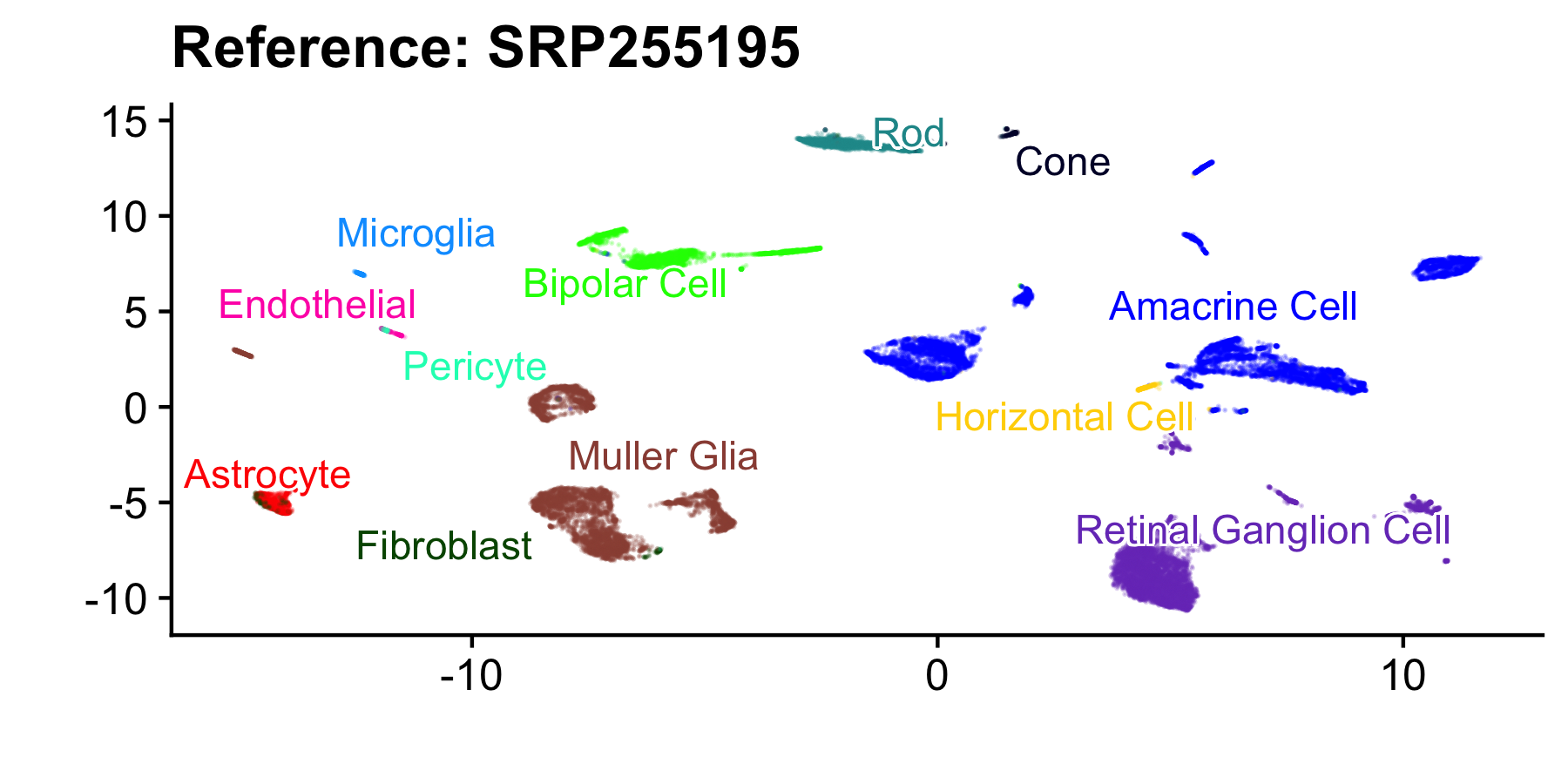
metamoRph::metamoRph
This takes the rotation matrix (prcomp’s $rotation) and
center/scale values calculated in metamoRph::run_pca and
the query (the new) data as input. The query data features (genes) are
matched to the reference data, normalized in the same manner as the
reference data, then multiplied against the rotation matrix. This
creates a sample by PCA space which can be directly compared with the
PCA of the reference data.
# https://hpc.nih.gov/~mcgaugheyd/scEiaD/2021_11_11/study_level/E-MTAB-7316.seurat.Rdata
load("~/Downloads/E-MTAB-7316.seurat.Rdata")
new_data <- scEiaD
pmat <- new_data@assays$RNA@counts
proj <- metamoRph(new_counts = pmat, rotation = mm_pca$PCA$rotation[, 1:50], center_scale = mm_pca$center_scale)
umap_new_data <- umap_transform(proj[, 1:20], umap_reference)
b <- umap_new_data %>%
as_tibble(rownames = "bc") %>%
left_join(bind_rows(reference@meta.data %>%
as_tibble(rownames = "bc"), new_data@meta.data %>%
as_tibble(rownames = "bc"))) %>%
filter(!is.na(CellType_predict), CellType_predict %in% celltypes_to_keep) %>%
ggplot(aes(x = V1, y = V2, color = CellType_predict)) + scattermore::geom_scattermore(pointsize = 2) +
cowplot::theme_cowplot() + xlab("") + ylab("") + scale_color_manual(values = man_color) + ggrepel::geom_label_repel(data = . %>%
group_by(CellType_predict) %>%
summarise(V1 = mean(V1), V2 = mean(V2)), aes(label = CellType_predict), alpha = 0.9) + theme(legend.position = "none") +
ggtitle("Morphed: EMTAB7316")
cowplot::plot_grid(a, b, ncol = 2, align = "v", axis = "r")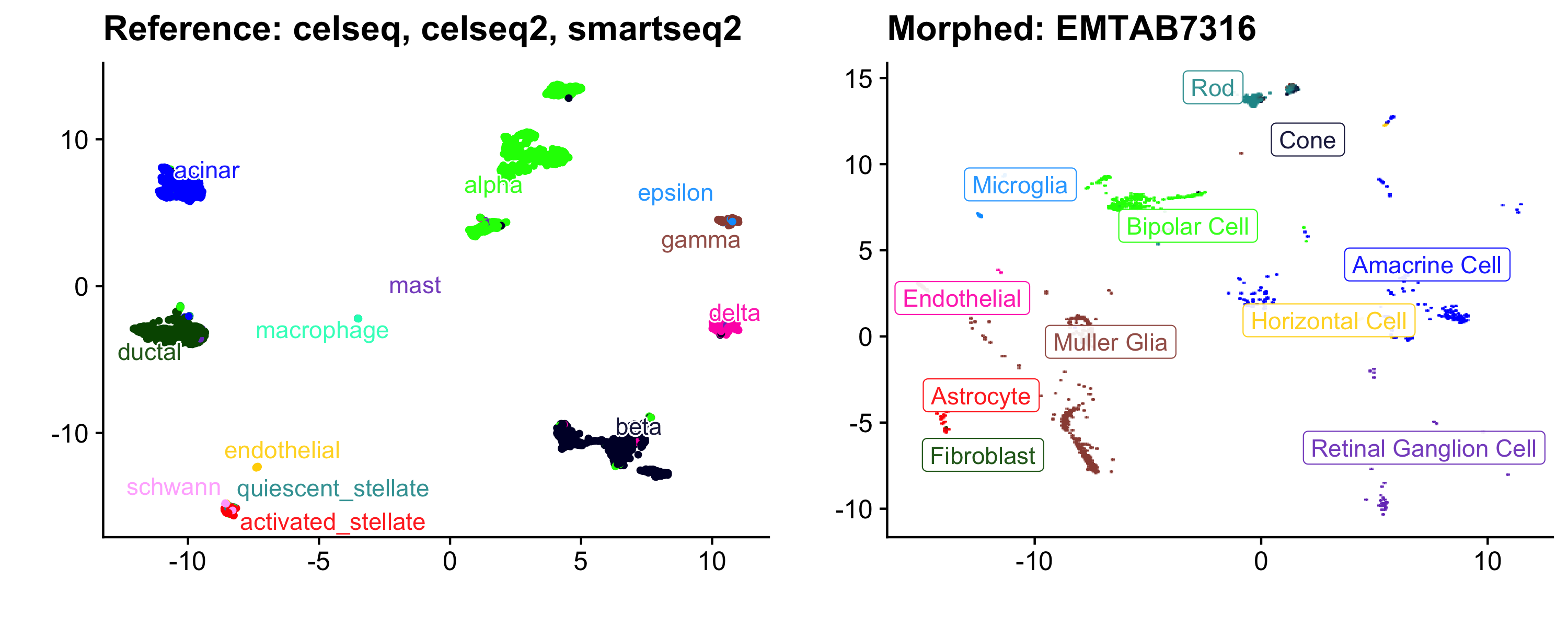
Label Transfer via ML
metamoRph::model_build
metamoRph also can perform label transfer (that’s the meta
in metamoRph) with metamoRph::model_build. It
trains a linear regression for each unique entry in the metadata field.
So if you have seven cell types, it will train seven models which are
tuned to distinguish each cell type against all remaining data.
You may be wondering why I’m using something as simple as a linear regression. Model build does support different models. Right now it also can use random forest, xgboost, glm, and svm. In practice I suggest the linear regression (“lr”) as it reliably performs the best and is the fastest option. SVM is a pretty close second. I have also experimented with several neural network based models and as they all perform substantially worse, I chose not to make them available.
metamoRph::model_apply
metamoRph::model_build returns a list object with a
model trained for each unique field (in this case cell types). This is
used directly in metamoRph::model_apply along with your
query / new data’s output from metamoRph::metamoRph. It
will return the predicted label along with the “max_score”, which is the
highest score returned each of the individual models. Closer to 0
indicates low confidence while 1 indicates high confidence.
As we already have labels for the query data we can also give
metamoRph::model_apply the known labels and the function
will output them with the guessed labels. That way you can quickly check
the accuracy, in this case it is near 98%.
ml <- metamoRph::model_build(mm_pca$PCA$x[, 1:50], mm_pca$meta %>%
pull(CellType_predict))
ma <- metamoRph::model_apply(ml, proj, new_data@meta.data$CellType_predict)
ma %>%
head()
#> # A tibble: 6 × 6
#> sample_id sample_label predict predict_second predict_stringent max_score
#> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 AAACCTGAGAGGTAGA_ERS2852885 Rod Rod Bipolar Cell Unknown 0.451
#> 2 AAACCTGAGCCATCGC_ERS2852885 Rod Rod Bipolar Cell Rod 0.517
#> 3 AAACCTGCAGACGTAG_ERS2852885 Rod Rod Muller Glia Unknown 0.464
#> 4 AAACCTGGTGCTGTAT_ERS2852885 Rod Rod Bipolar Cell Unknown 0.462
#> 5 AAACCTGTCAAGATCC_ERS2852885 Amacrine Cell Amacrine Cell Pericyte Amacrine Cell 1.53
#> 6 AAACCTGTCTTGAGAC_ERS2852885 Horizontal Cell Horizontal Cell Amacrine Cell Horizontal Cell 0.928
# overall accuracy
ma %>%
filter(sample_label == predict) %>%
nrow()/ma %>%
filter(!is.na(sample_label)) %>%
nrow()
#> [1] 0.984965